
80 % of PDAC is made of desmoplastic stroma
At diagnosis of pancreatic ductal adenocarcinoma, the median overall survival with the best multi-agent
regimens still hovers around eleven months. The five-year relative survival rate has crept from 3 percent
in the 1970s to roughly 13 percent in 2025.
Why has pancreatic cancer resisted the transformations that reshaped the rest of oncology ?
The defining histological feature of pancreatic ductal adenocarcinoma (PDAC) is desmoplasia: a dense,
fibrotic reaction in which extracellular matrix and stromal cells crowd out the malignant epithelium. In a
typical PDAC biopsy, 80 to 90 percent of what you see under the microscope is not cancer cells at all. It is
collagen, hyaluronic acid, activated fibroblasts, suppressed immune cells, and a tangle of signalling
molecules. The cancer cells themselves are a minority population, embedded in a tissue of their own creation.
The fortress metaphor is more than decorative. Three barrier functions explain much of the disease’s
clinical behaviour.
First, the high concentration of hyaluronic acid traps fluid and raises interstitial pressure to levels that
physically collapse the tumour’s blood vessels. Chemotherapy delivered intravenously cannot reach cells
whose vasculature has been crushed.
Second, the matrix walls off the tumour from the immune system, both physically and chemically. T cells
that might recognise and destroy malignant cells cannot penetrate the dense fibrotic envelope, and the
chemokine architecture of the stroma actively repels them a point we will return to in detail.
Third, and most subtly, the matrix is not inert. Extracellular matrix proteins actively transmit survival
signals to the cancer cells inside, promoting epithelial-to-mesenchymal transition, chemoresistance, and
stem-cell-like behaviour.
Behind all of this stands a single master regulator: TGF-β. This cytokine recruits and activates the
fibroblasts that lay down the collagen, orchestrates the immunosuppressive milieu, and drives the
epithelial-to-mesenchymal transition that lets cancer cells escape the primary tumour. TGF-β has its own
paradox early in carcinogenesis it suppresses tumour formation; once the tumour is established, the
same molecule becomes its most reliable accomplice. This duality is the signature of the pancreatic
stroma itself.
From this picture an obvious therapeutic strategy emerged in the early 2000s, with the clarity of a
syllogism. The stroma blocks treatment. Therefore, dissolve the stroma. The drugs will then reach their
target, the immune system will rejoin the fight, and survival will improve.
Two decades of trials would test this proposition. Almost all of them failed.
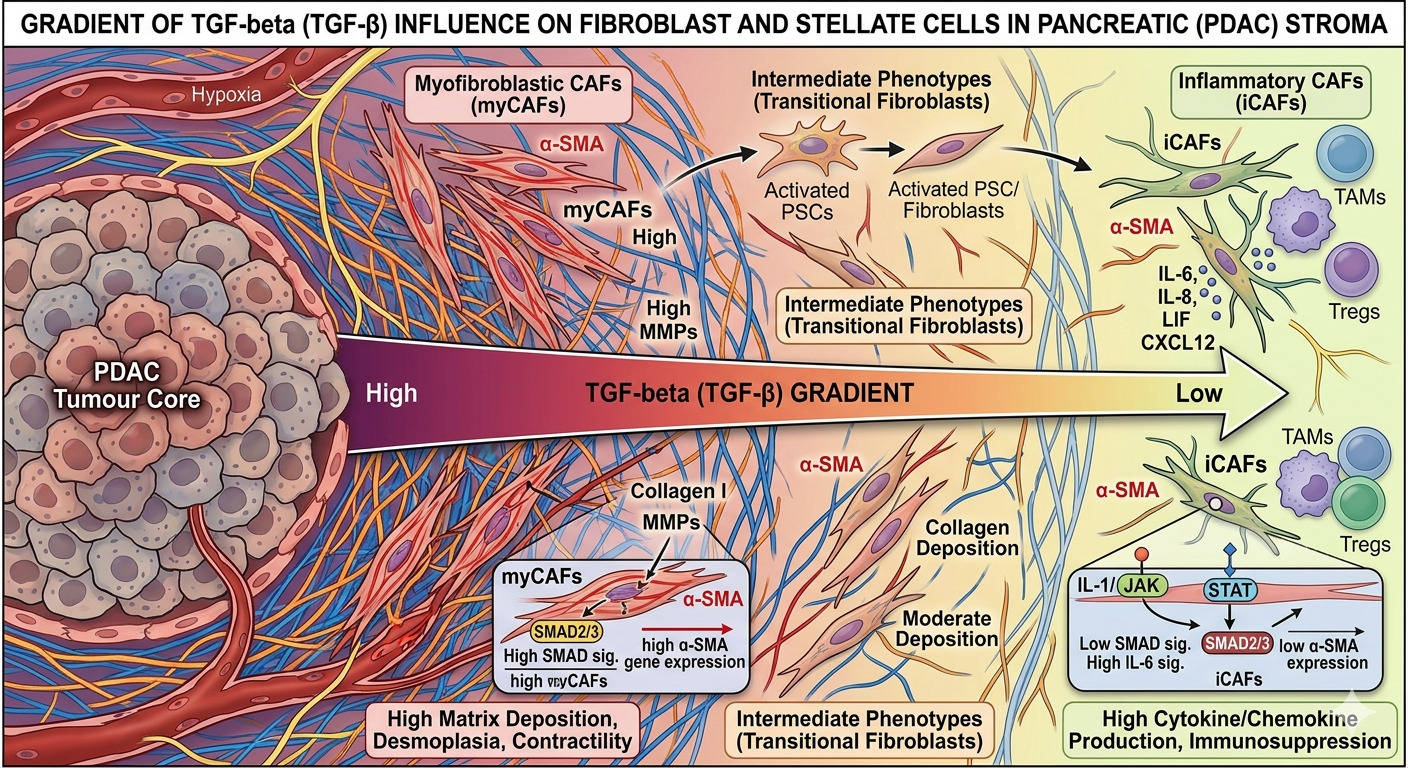
Influence of TGF beta gradient over fibroblast subtypes transformation
picture generated by AI
Failure of immune checkpoint blockade therapy in PDAC
PDAC is the prototype “cold” tumour. Its mutational burden is low, generating few neoantigens. Its
KRAS-mutant epithelium downregulates MHC class I, hiding what neoantigens it does produce. And its
microenvironment is built to keep effector T cells out and suppressor cells in.
The cellular composition of the PDAC immune infiltrate is itself a barrier. Tumour-associated
macrophages (TAMs), recruited largely through the CSF1/CSF1R axis, polarise toward an M2-like
phenotype that secretes IL-10, IL-6, and TGF-β. Myeloid-derived suppressor cells (MDSCs) both
granulocytic and monocytic are recruited by CXCL1, CXCL2, CXCL5, and GM-CSF, and suppress T cell
responses through arginase-1, inducible nitric oxide synthase, and reactive oxygen species. Regulatory T
cells (Tregs) expanded by local TGF-β contribute IL-10 and IL-35. Together these populations dominate
the lymphocytic compartment from the earliest precursor lesions onward, before any frank carcinoma
has formed.
The few effector T cells that do enter the tumour are typically held at the periphery by a chemokine
cloak CXCL12 secreted by FAP-positive stromal fibroblasts coats the malignant epithelium and excludes T cells from contact with the cancer cells they would otherwise kill. When effector cells do
penetrate, they are characteristically dysfunctional, expressing not only PD-1 but a panel of
compensatory checkpoints TIGIT, TIM-3, LAG-3 any of which can sustain exhaustion when one is
blocked.
The clinical correlate is unambiguous. Single-agent anti-PD-1 and anti-PD-L1 antibodies are essentially
inactive in microsatellite-stable PDAC, which is to say, in 97 to 99 percent of cases. The dual checkpoint
blockade combination of tremelimumab and durvalumab produced an objective response rate of 3.1
percent in advanced PDAC, and adding it to gemcitabine plus nab-paclitaxel produced no survival benefit
over chemotherapy alone. The single approval that exists pembrolizumab for mismatch-repair-
deficient or microsatellite-instability-high tumours, granted in 2017 on a tissue-agnostic basis covers
fewer than one percent of PDAC patients and even in that group produced an objective response rate of
only 18 percent and a median overall survival of 4 months.
This is the immune problem the field is now trying to solve. As we will see, the most encouraging recent
strategies do not attempt to bypass the stromal barrier; they work through it.
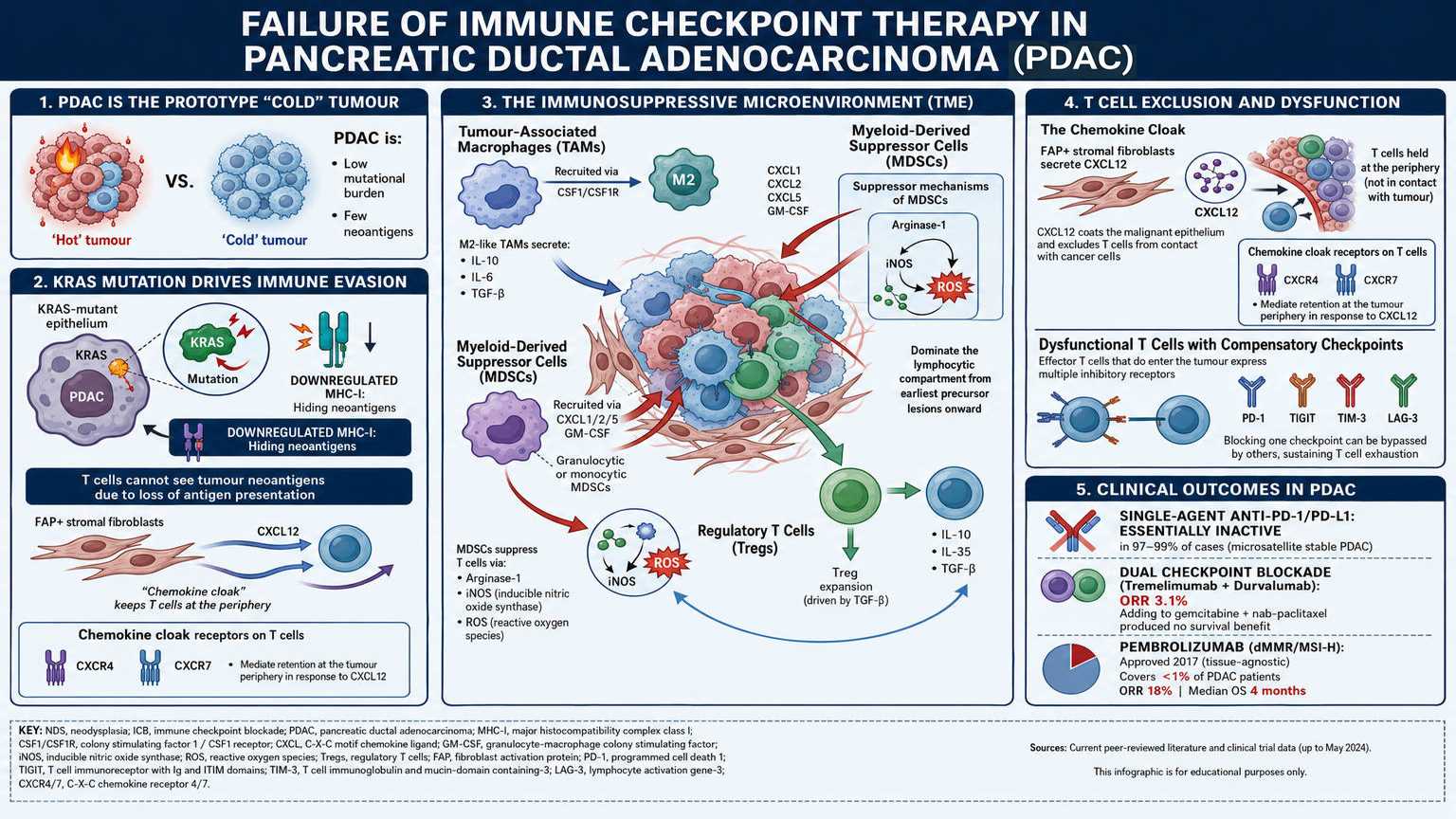
Failure of immune checkpoint blockade therapy in PDAC
picture generated by AI
The trials that should have worked : PEGPH20
PEGPH20 and the delivery hypothesis
Of all the strategies tried, PEGPH20 had the cleanest logic. Pegvorhyaluronidase alfa is a pegylated
recombinant human hyaluronidase that degrades hyaluronic acid, the gel-like polysaccharide
responsible for much of the interstitial pressure in PDAC. The reasoning was direct: degrade the
hyaluronan, lower the pressure, restore vascular flow, get gemcitabine and nab-paclitaxel to the cancer
cells. Preclinical mouse data were striking. Early-phase trials selecting hyaluronan-high tumours
suggested benefit. The FDA granted orphan drug designation.
The pivotal phase III trial randomised 492 patients with hyaluronan-high metastatic PDAC, two-to-one,
between PEGPH20 plus standard chemotherapy and chemotherapy alone. The primary endpoint was
overall survival. The result, reported at the 2020 Gastrointestinal Cancers Symposium and published in
the Journal of Clinical Oncology that same year: 11.2 months in the experimental arm versus 11.5
months in the control arm, with a hazard ratio of 1.00 and a p-value of 0.97. The trial was a complete
null result, and Halozyme halted all further development. A parallel SWOG cooperative-group study
testing PEGPH20 with FOLFIRINOX rather than gemcitabine plus nab-paclitaxel was equally negative. 6
Why did it fail? Two reasons, mechanistically connected. The PEGPH20 arm suffered substantially more
toxicity, including thromboembolic events severe enough that prophylactic enoxaparin had been added
during the trial design. Patients on the experimental arm received fewer cycles of chemotherapy and
underwent more dose reductions. The drug worked on the stroma response rates were higher but
the patients did worse, because they could not tolerate enough chemotherapy to translate that
response into survival.
More fundamentally, even when delivery improved, the cancer cells had other escape routes: epithelial-
to-mesenchymal transition, low mutational burden, paucity of neoantigens. Removing the wall did not,
by itself, make the prisoners surrender.
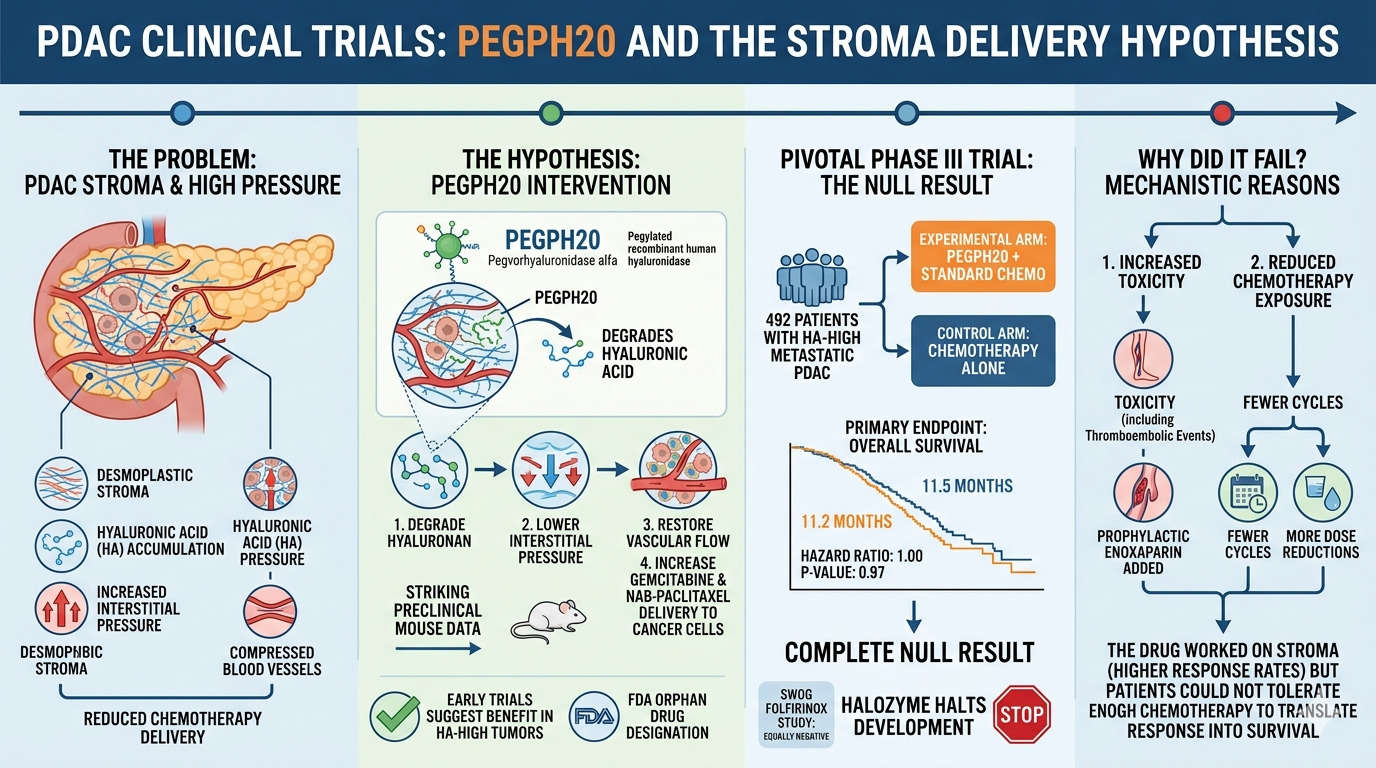
PEGPH20 and the delivery hypothesis
picture generated by AI
another failure : approach of Sonic Hedgehog pathway and Saridegib, vismodegib depletion hypothesis
The Sonic Hedgehog pathway drives the
differentiation of pancreatic stellate cells into the activated fibroblasts that build the desmoplastic
reaction. Block SHH signalling and the stroma should not just thin it should disintegrate.
The drug chosen for the first phase II trial had a remarkable origin. In the 1950s, sheep ranchers in Idaho
began noticing newborn lambs with grotesque craniofacial deformities most strikingly, a single
central eye. The pattern was epidemic in some flocks. Agricultural investigators eventually traced the
cause to pregnant ewes grazing on the corn lily, Veratrum californicum. The plant contained a steroidal
alkaloid that, ingested at a critical window of gestation, produced cyclopia. The compound was named
cyclopamine. Decades later, molecular biology revealed that cyclopamine was a teratogen because it
inhibited Hedgehog signalling a pathway essential to the patterning of the vertebrate embryo. A
poisonous plant on a Western rangeland had revealed an entire developmental signalling system.
Saridegib (IPI-926) is a semi-synthetic derivative of cyclopamine. The therapeutic logic was elegant: a
molecule that disrupts tissue organisation in embryos might disrupt the aberrant tissue organisation of a
tumour. Preclinical work in genetically engineered KPC mice was spectacular. Saridegib depleted the
stroma, increased tumour vascular density, improved gemcitabine delivery, and prolonged survival.
The randomised phase II trial run by Infinity Pharmaceuticals (NCT01130142) compared gemcitabine
plus saridegib with gemcitabine plus placebo in metastatic PDAC. It was halted at interim analysis in
January 2012. Patients receiving the Hedgehog inhibitor had worse progression-free survival and worse
overall survival than those receiving placebo. They were dying faster. Development of saridegib for
pancreatic cancer was abandoned.
A second Hedgehog inhibitor vismodegib (GDC-0449), already approved for basal cell carcinoma
was tested in a parallel randomised phase Ib/II trial led by Catenacci and reported in the Journal of
Clinical Oncology in 2015. Adding vismodegib to gemcitabine made no significant difference in either
progression-free or overall survival. 7 A subsequent phase II study combining vismodegib with
gemcitabine plus nab-paclitaxel likewise failed to improve outcomes over historical controls. 8 Hedgehog
inhibition in established PDAC was definitively closed as a strategy.
The reckoning was uncomfortable. The drugs had done exactly what they were supposed to do: they
had depleted the stroma. The problem was that depleting the stroma had unleashed the tumour. Two
landmark mouse studies published in Cancer Cell in 2014 by the Kalluri group (Özdemir and
colleagues) and the Stanger group (Rhim and colleagues) showed why. When αSMA-positive
myofibroblasts were genetically deleted from PDAC, the tumours that resulted were not smaller and
slower; they were more aggressive, more poorly differentiated, more metastatic, and survival in the
mice was reduced. The stroma had been doing two things simultaneously. It had been blocking drug
delivery the failure mode the field had focused on and it had been physically and biochemically
restraining the cancer cells. The fortress walls had been keeping the prisoners in. Once they came down,
the most aggressive clones escaped.

Cyclopamine interrupts the sonic hedgehog signalling pathway : teratogen but potential drug
picture generated by AI, close to lesions found in some ranch in Idaho in the fifties showing plant Veratrum californicum and consequence on cattle
The Failure of matrix-remodelling hypothesis : Marimastat and simtuzumab examples
The matrix metalloproteinase inhibitor marimastat, designed to block
the proteolytic remodelling that supports invasion, performed worse than gemcitabine alone in the
randomised trial reported by Bramhall in 2002. 9 Simtuzumab, a humanised antibody targeting LOXL2
an enzyme that crosslinks collagen and was correlated with PDAC aggressiveness added no survival
benefit to gemcitabine in the phase II randomised trial reported by Benson in 2017. 10 Each trial targeted
a different stromal component. Each failed.
Losartan : old drug and an unexpected asset
One striking partial exception emerged from a different premise. Rakesh Jain and colleagues at
Massachusetts General Hospital had shown in mouse models that the angiotensin II type 1 receptor
blocker losartan already used for decades as an antihypertensive inhibits collagen I production by
activated fibroblasts, decompresses tumour vasculature, and improves drug and oxygen delivery
without depleting the stromal cell populations. 11 In other words, losartan modulates the matrix rather
than dismantling its cellular machinery.
A single-arm phase II trial led by Murphy and colleagues (NCT01821729) added losartan to FOLFIRINOX
and chemoradiation in 49 patients with locally advanced pancreatic cancer. The results, published in
JAMA Oncology in 2019, were striking: an R0 resection rate of 61 percent historically unattainable in
this population and a median overall survival of 31.4 months from enrolment. 12 Circulating TGF-β fell
during treatment, consistent with effective stromal modulation.
The caveats are real: it was a single-arm study in a selected patient population, surgical and radiation
expertise contributed substantially, and a randomised confirmation has not yet been completed. The
point is conceptual rather than evidentiary. Losartan worked, where saridegib failed, because it did not
try to demolish the stroma. It rebalanced it.
By the late 2010s the pattern was unmistakable. Hyaluronan, Hedgehog, matrix metalloproteinases, lysyl
oxidase, αSMA-positive myofibroblasts: every attempt to identify a single stromal target and remove it
had either produced no benefit or made patients worse. The error, the field was forced to concede, was
not in the choice of target. It was in the assumption that the stroma was a single thing.
The stroma is not monolithic
The conceptual breakthrough came from single-cell biology. When researchers stopped treating the
PDAC stroma as a homogeneous mass and began profiling its individual cells, the picture transformed.
Work led by David Tuveson’s group at Cold Spring Harbor first by Öhlund and colleagues in 2017, then
extended by Elyada and colleagues in 2019 revealed that the cancer-associated fibroblasts (CAFs)
within a single PDAC are not a uniform population. They comprise at least three distinct subtypes with
opposing functions, intermingled within the same tumour. 13
Myofibroblastic CAFs (myCAFs) sit close to the cancer cells, are activated by direct contact and TGF-β
signalling, and express high levels of αSMA. They are the wall-builders. They lay down collagen, organise
the matrix, and physically constrain the tumour. The 2014 mouse experiments by Özdemir and Rhim
showed that depleting these cells accelerates disease these are the fibroblasts whose restrictive
function had been silently doing the work the chemotherapy could not.
Inflammatory CAFs (iCAFs) sit further from the malignant epithelium, are activated by paracrine IL-1
signalling through JAK/STAT, and secrete a stew of inflammatory cytokines including IL-6. They are not
wall-builders but collaborators. They actively support tumour growth, drive chemoresistance, and
contribute to the immunosuppressive environment. They are precisely the population one would want
to suppress.
Antigen-presenting CAFs (apCAFs), characterised more recently, express MHC class II and can present
antigen to CD4+ T cells. Whether they are net protective, net suppressive, or context-dependent in
human PDAC remains contested.
Critically, these populations are interconvertible. TGF-β pushes fibroblasts toward the myCAF state; IL-1
and JAK/STAT signalling pushes them toward the iCAF state. The balance is dynamic. Deplete one
population indiscriminately and the others compensate.
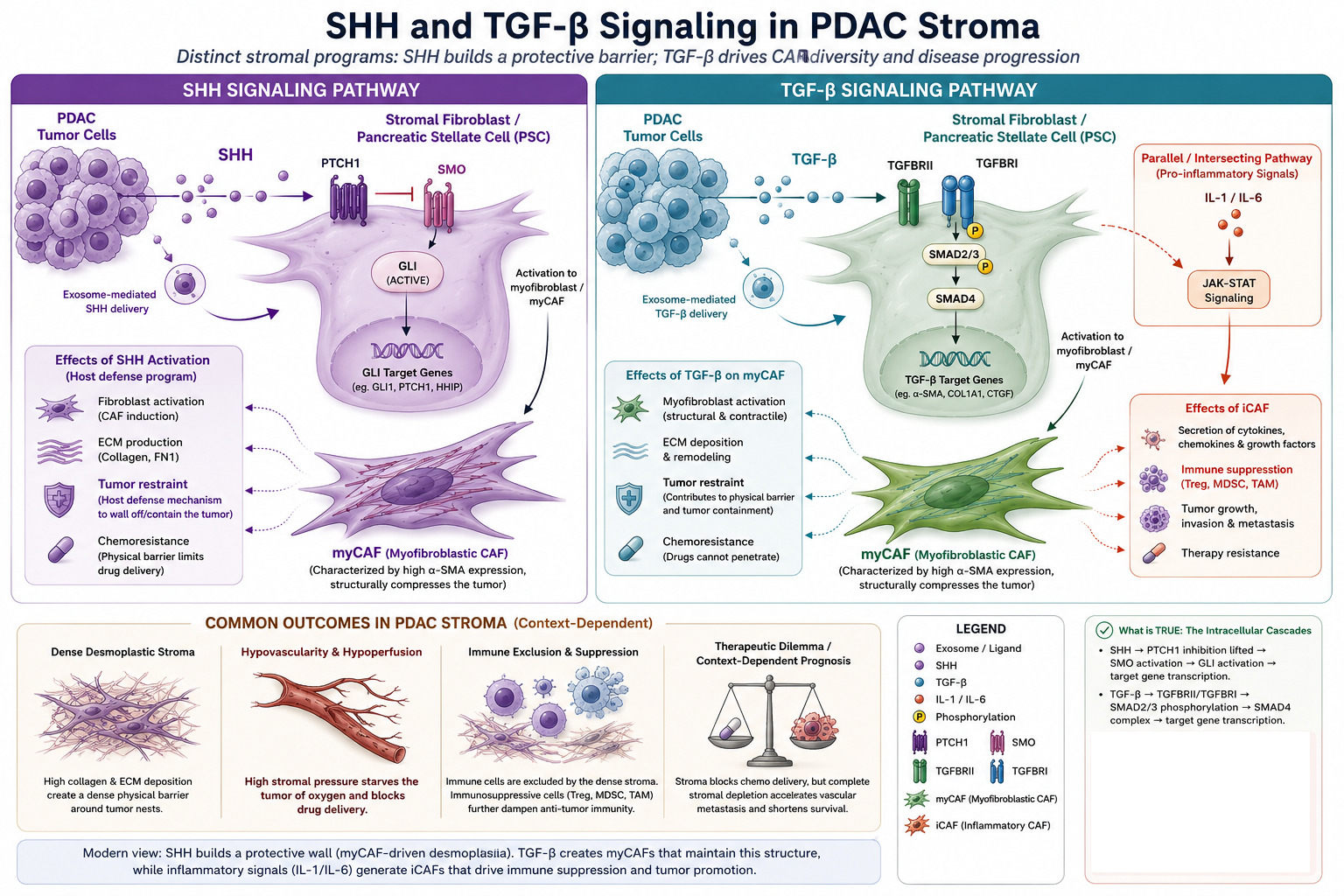
Summurize of SHH and TGF beta roles
picture generated by AI
The exosome highway
The stroma is not only heterogeneous. It is communicative and the language is largely vesicular. Cells
in the PDAC microenvironment exchange a continuous traffic of small extracellular vesicles 30 to 150
nanometres in diameter, derived from endosomal multivesicular bodies and bounded by membranes
enriched in tetraspanins (CD9, CD63, CD81) and ESCRT components (ALIX, TSG101). These exosomes
carry curated cargoes of proteins, lipids, microRNAs, mRNAs, long non-coding RNAs, and double-
stranded DNA fragments protected from extracellular nucleases and selectively taken up by recipient
cells. 15
Three vectors of crosstalk matter clinically.
CAF-to-tumour exosomes drive chemoresistance. Exposure to gemcitabine itself triggers CAFs to release
exosomes loaded with PTEN-targeting microRNAs miR-21, miR-181a, miR-221, miR-222, and miR-92a
which suppress PTEN in recipient PDAC cells and induce gemcitabine resistance. 16 In other words, the
chemotherapy that fails to reach the cancer cells provokes the stroma to broadcast the resistance signal.
Tumour-to-tumour exosomes spread chemoresistance horizontally. PDAC cells under prolonged
gemcitabine exposure upregulate miR-155, which both promotes anti-apoptotic signalling and increases
exosomal miR-155 secretion. The miR-155 cargo, transferred to neighbouring cells, suppresses
deoxycytidine kinase the enzyme that activates gemcitabine and propagates resistance through
the tumour mass. 17 Gemcitabine-resistant PDAC stem cells similarly transfer miR-210 to sensitive cells,
activating PI3K/AKT/mTOR signalling and inducing the resistance phenotype.
Tumour-to-stellate-cell exosomes are where the desmoplastic reaction is sustained. Tumour-derived
exosomes carrying TGF-β and other factors activate quiescent pancreatic stellate cells into the CAF
phenotype, completing a feedback loop that maintains the stroma even as it remodels. The same
vesicular traffic shapes immune cells: PDAC exosomes enriched in arachidonic acid and bearing surface
ICAM-1 fuse preferentially with macrophages and contribute to their M2 polarisation. 18 Tumour
exosomes bearing macrophage migration inhibitory factor (MIF) reach the liver via the portal circulation
and prime hepatic Kupffer cells to form the pre-metastatic niche before any macrometastatic disease
has developed. 19
Drugs like saridegib did not distinguish between myCAFs and iCAFs; they hit both. PEGPH20 dissolved a
substrate that all CAF populations contributed to without addressing their distinct biological roles. None
of the trials touched the exosomal communication system that maintains the whole arrangement. We
were treating a complex, dynamic ecosystem as if it were a uniform tissue, and applying demolition
charges where what was needed was selective negotiation.
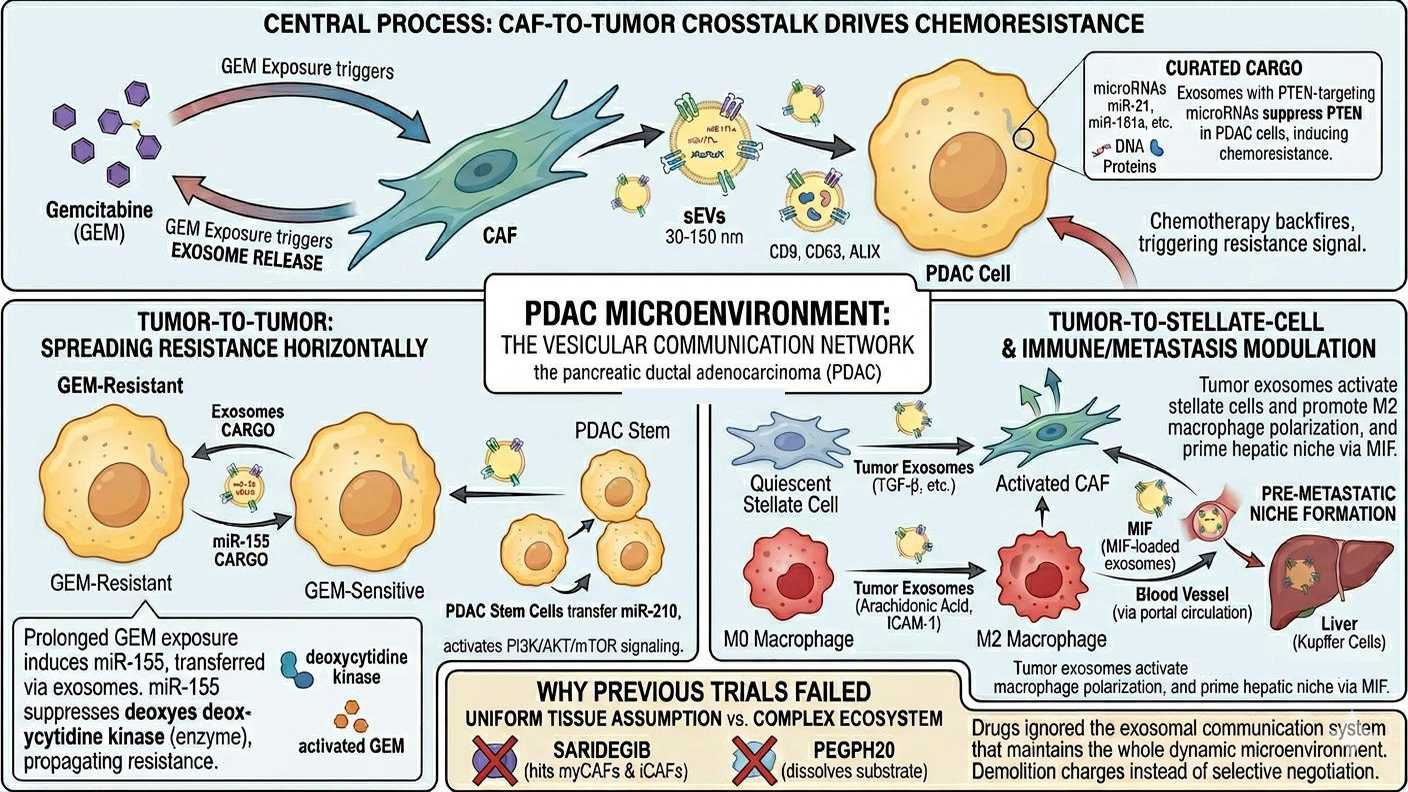
Microvesicles communication between stroma and tumoral cells
picture generated by AI
The CXCL12 axis and the CXCR7 problem
If a single signalling axis ties together the stromal, immune, and chemotactic features of PDAC, it is
CXCL12 and its receptors. CXCL12 (also known as SDF-1) is secreted in large quantities by FAP-positive
stromal fibroblasts. Coating the tumour epithelium, it both excludes T cells from the malignant cells and
recruits suppressive myeloid populations into the microenvironment. As Feig and colleagues had shown
in 2013, depleting FAP-positive stromal cells, or pharmacologically inhibiting CXCR4, reverses T cell
exclusion and synergises with anti-PD-L1 therapy in mouse PDAC the experimental foundation for the
clinical strategy that followed.
The resulting clinical strategy combined the CXCR4 antagonist motixafortide (formerly BL-8040) with
pembrolizumab and chemotherapy. The phase IIa COMBAT/KEYNOTE-202 trial enrolled patients with
metastatic PDAC progressing after first-line gemcitabine-based therapy. In the initial Bockorny Nature
Medicine report in 2020, the triple-combination cohort of 22 patients showed an objective response
rate of 32 percent and a disease control rate of 77 percent startling figures in a disease essentially
refractory to checkpoint blockade. 20 The expanded cohort of 43 patients reported by Bockorny in
Clinical Cancer Research in 2021 produced more conservative numbers an objective response rate of
21 percent (confirmed 13 percent), a disease control rate of 63 percent, and a median overall survival of
6.6 months but the immunological signal held: motixafortide measurably increased intratumoural
CD8+ T cell infiltration and decreased circulating MDSCs and Tregs. 21 A randomised phase II trial is now
underway.
It was at this point that a quieter problem came back into focus. CXCL12 has not one receptor but two:
CXCR4 and CXCR7 (also known as ACKR3, atypical chemokine receptor 3). PDAC cells co-express both,
and the two have different signalling logics. CXCR4 is a conventional G-protein-coupled receptor; CXCR7
is a β-arrestin-coupled scavenger that internalises and degrades CXCL12 but can also redirect signalling
through MAPK and Akt pathways. Plerixafor (AMD3100), the first-generation CXCR4 antagonist, was
shown to paradoxically activate CXCR7, undermining the very blockade it was meant to deliver. 22
Gemcitabine itself upregulates both CXCR4 and CXCR7 in PDAC cells, providing one mechanism for
acquired chemoresistance.
This receptor duality is one reason CXCR4-only blockade may be incomplete. It is also why ligand-
targeted strategies such as the CXCL12-binding aptamer NOX-A12 (olaptesed pegol), which
neutralises CXCL12 itself and therefore disables both receptors and dual CXCR4/CXCR7 antagonists
are being actively developed. Whether motixafortide’s clinical signal will hold up against this
incompleteness remains an open question; the next round of randomised data should clarify it.
From demolition to reprogramming
The new generation of stromal therapy reflects the lessons of the failed era. The goal is no longer to
dissolve the fortress but to reprogram its inhabitants silencing the iCAFs and their inflammatory
output, preserving the restrictive myCAFs, breaking specific communication channels rather than
dismantling the entire structure, and combining stromal modulation with immune activation rather than
expecting either to work alone.
Several strategies follow this logic.
Targeting focal adhesion kinase (FAK), which links cancer cells to the stromal matrix, reduces immune
exclusion through a mechanism distinct from CXCR4 blockade. The Nature Medicine paper by Jiang and
colleagues in 2016 showed that FAK inhibition in mouse PDAC reduces fibrosis and Treg infiltration and
renders previously resistant tumours responsive to checkpoint immunotherapy. 23 An early-phase trial of
the FAK inhibitor defactinib combined with pembrolizumab and gemcitabine produced modest activity
one partial response and seven instances of stable disease in ten evaluable patients and confirmed
tolerability. 24 Several later-phase studies are ongoing, including a randomised phase II combining
defactinib and avutometinib with stereotactic body radiotherapy.
TGF-β inhibition is being revisited in selective form rather than as global pathway suppression.
Galunisertib plus gemcitabine produced a modest survival signal in a 2018 phase II trial. 25 Bifunctional
anti-PD-L1/TGF-β agents have been more disappointing bintrafusp alfa was discontinued in non-small
cell lung cancer in 2021 but the principle of pairing checkpoint blockade with TGF-β neutralisation in
subtype-stratified PDAC populations remains active.
KRAS-directed immunotherapy has produced the most encouraging early signal in years. A pooled
synthetic long peptide vaccine targeting the six common KRAS mutations (G12D, G12V, G12R, G12C,
G12A, G13D) combined with ipilimumab and nivolumab was tested by the Jaffee group in resected
PDAC. In the phase I trial reported by Huff and colleagues in 2026, the combination was safe and
induced antigen-specific T cell responses in 11 of 12 patients, with cross-reactivity across multiple KRAS
mutants. 26 Whether immunogenicity translates into recurrence-free survival benefit will require
randomised data, but the strategy directly attacks both halves of the immune problem neoantigen
poverty and checkpoint dependence and does so against a target shared by more than 90 percent of
PDAC.
Several trials are now stratifying patients by stromal subtype myCAF-rich versus iCAF-rich tumours
appear to behave differently and may respond to different combinations.
None of this has yet produced a transformative survival benefit. The real test will come over the next
several years, as randomised phase III data emerge from the motixafortide, FAK, KRAS-vaccine, and
losartan programmes. But the conceptual frame has shifted decisively. The stroma is no longer treated
as an obstacle to be removed but as a tissue to be modulated and its immune compartment, far from
being a bystander, is now the principal lever of progress.